Molecular dynamics (MD) simulations are an essential tool in computational biology, chemistry, and materials science. They allow scientists to model and predict the behavior of molecular systems at an atomic level over time. From understanding protein folding to exploring drug-receptor interactions, MD simulations provide unparalleled insight into molecular systems. However, traditional MD simulations are notoriously computationally expensive and can require weeks of processing to simulate just a few nanoseconds of real-world time.
This is where deep learning steps in. By combining accelerating MD simulation with deep learning, researchers can accelerate the simulation process, making it more efficient while maintaining accuracy. The integration of these two technologies is not only transforming how we approach molecular simulations but also opening new possibilities in fields like drug discovery and materials design.
In this article, we will explore the methods behind accelerating MD simulation with deep learning and dive into key applications that are pushing the boundaries of scientific research.
The Basics of Molecular Dynamics (MD) Simulations
Molecular dynamics simulations are based on Newton’s laws of motion, which are used to calculate the movement of atoms and molecules over time. These simulations require:
- Initial atomic positions: The starting configuration of atoms in a molecule or molecular system.
- Force fields: Mathematical models that describe the forces acting on each atom based on their interactions.
- Time integration: Repeated calculations to update atomic positions and velocities over time, producing a trajectory of the molecular system.
MD simulations can offer detailed insights into molecular behaviors, but they are computationally intensive. For large molecules like proteins or complex systems like drug-receptor interactions, MD simulations can take weeks or months to run on even the most advanced hardware.
Limitations of Traditional MD Simulations
- High Computational Cost: The sheer number of atoms involved, coupled with the need to calculate every interaction at each time step, makes MD simulations very slow.
- Limited Timescales: While many biological processes occur over milliseconds or longer, traditional MD simulations often can only handle nanosecond or microsecond timescales.
- Scaling Challenges: Simulating large systems with thousands or millions of atoms quickly becomes unmanageable due to the exponential growth in computational requirements.
Deep Learning as a Solution
Deep learning is a subset of machine learning that uses artificial neural networks to recognize patterns and make predictions based on large amounts of data. By leveraging deep learning, researchers can predict the behavior of molecular systems without the need to compute every atomic interaction at every time step, significantly speeding up the simulation process.
Accelerating MD Simulation with Deep Learning – How it Works?
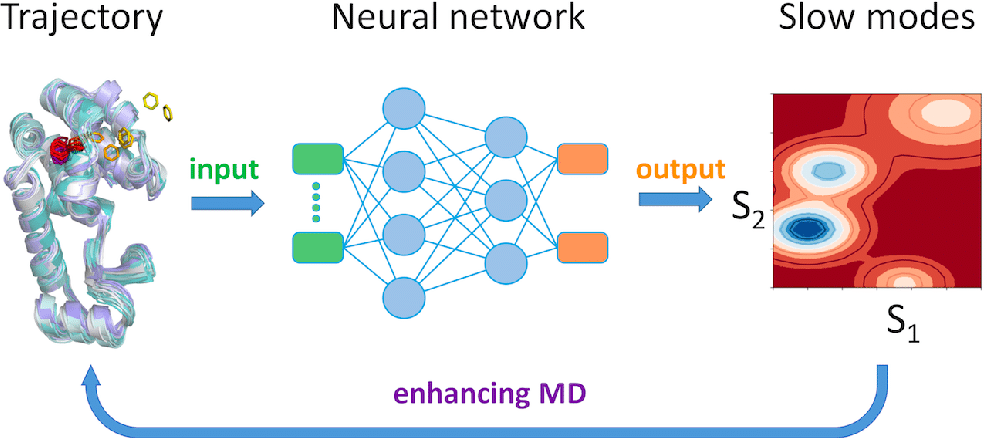
1. Predicting Molecular Dynamics Using Neural Networks
One of the primary ways accelerating MD simulation with deep learning is accelerating research is through the use of neural networks to predict molecular behavior. These models are trained on data from traditional MD simulations, learning to predict the future state of a molecular system based on its current state.
For example, time-lagged autoencoders (TAEs) are neural networks that learn how molecular configurations evolve over time. Once trained, these models can predict future configurations without needing to calculate every intermediate step, drastically reducing the computational load.
2. Learning Force Fields
Force fields are the cornerstone of MD simulations, determining how atoms in a system interact with each other. Traditional force fields are often based on simplified physical models that can introduce inaccuracies, especially in complex systems.
Deep learning offers an alternative: neural network potentials (NNPs). These are deep learning models that are trained on high-accuracy quantum mechanical data and can predict atomic interactions more precisely than traditional force fields. By learning from massive datasets of atomic configurations, NNPs can generalize to predict the interactions in new, unseen systems, leading to more accurate simulations.
3. Generating Long-Timescale Dynamics
Many biological processes, such as protein folding, happen over timescales that are too long for traditional MD simulations to capture. To bridge this gap, deep learning can predict long-timescale behaviors from short-time simulations.
For instance, Markov state models (MSMs) break down the dynamics of a molecular system into a series of states and transitions. Deep learning can be used to predict these transitions and simulate how a molecular system will behave over long periods, allowing researchers to study processes that occur over milliseconds or longer, something traditional MD simulations cannot achieve without significant computational costs.
4. Reducing Sampling Errors
MD simulations often suffer from sampling inefficiencies, meaning that important molecular configurations may be missed because the simulation only explores a fraction of the possible states. Reinforcement learning, a branch of machine learning, is being applied to guide MD simulations toward the most relevant configurations, improving sampling efficiency and reducing the amount of computational time needed to explore important molecular behaviors.
5. Real-Time Simulations
In some applications, particularly drug discovery, researchers need real-time feedback on how a molecular system is behaving. Deep learning models integrated with MD simulations can predict molecular dynamics in real-time, allowing researchers to interact with the simulations and make decisions based on immediate results. This is especially useful for exploring drug-protein binding interactions and optimizing lead compounds in drug discovery pipelines.
Key Applications of Accelerating MD Simulation with Deep Learning
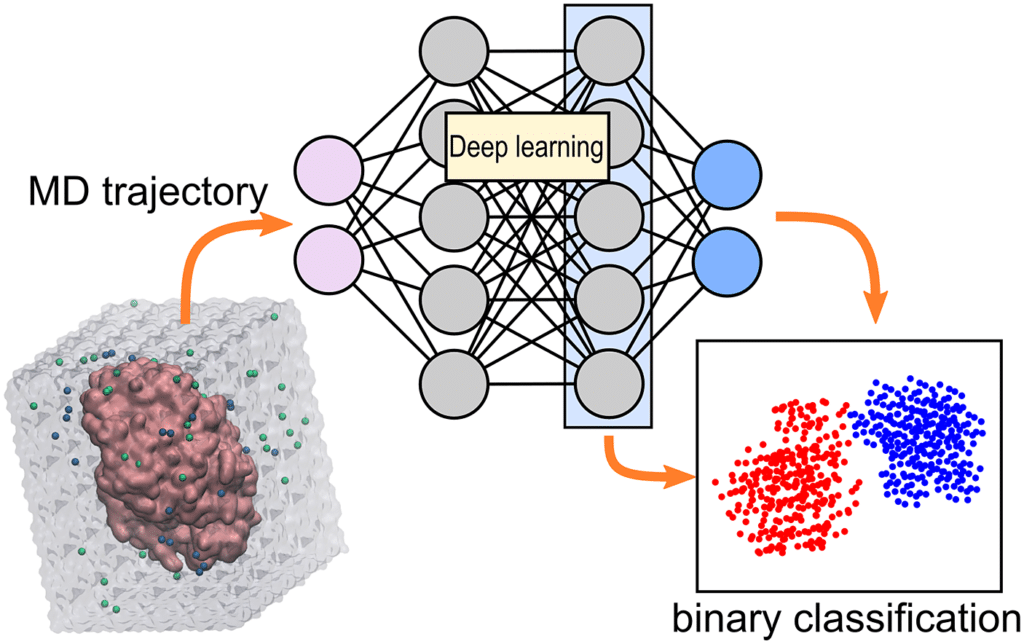
1. Drug Discovery
One of the most impactful applications of Accelerating MD simulation with deep learning is in drug discovery. Drug discovery involves identifying molecules that can bind to biological targets, typically proteins, to alter their function in a way that treats a disease.
Traditional MD simulations are often used to explore how drug molecules interact with proteins at an atomic level, but these simulations are limited by their computational expense. By incorporating deep learning, researchers can:
- Accelerate virtual screening: Deep learning models can quickly predict which drug candidates are most likely to bind to a target protein, reducing the need for full-scale MD simulations for every compound.
- Predict drug-protein binding: Deep learning-enhanced MD simulations provide faster and more accurate predictions of drug binding poses, crucial for optimizing drug candidates.
- Model allosteric sites: Allosteric sites are secondary binding locations on proteins that can modulate their activity. MD simulations augmented by deep learning can help identify and explore these sites, providing new avenues for drug design.
2. Protein Folding and Misfolding
Understanding how proteins fold into their functional forms is critical for drug design, especially for diseases caused by protein misfolding, like Alzheimer’s or Parkinson’s. However, simulating the full folding process is difficult because it can take milliseconds or longer, far beyond the reach of traditional MD simulations.
Deep learning models trained on MD simulation data can predict folding pathways and timescales, making it possible to study protein folding at a fraction of the computational cost. This has significant implications for developing drugs that can prevent or correct protein misfolding.
3. Materials Science
Beyond biology and drug discovery, accelerating MD simulation with deep learning is being applied in materials science to accelerate the discovery of new materials with desirable properties. For example, deep learning models can predict how atomic structures will behave under different conditions, allowing researchers to explore new materials for applications like batteries, semiconductors, or catalysts.
4. Biophysics and Structural Biology
In structural biology, MD simulations are used to study how biological macromolecules like proteins, DNA, and RNA interact. With deep learning, researchers can model larger systems and longer timescales, providing deeper insights into molecular mechanisms that are critical for understanding biological processes and disease.
Challenges and Future Directions
While the integration of accelerating MD simulation with deep learning has shown immense promise, there are still challenges to overcome:
- Data requirements: Training deep learning models requires large datasets, and generating high-quality data from MD simulations or quantum mechanical calculations can be computationally expensive.
- Model generalization: Deep learning models trained on specific molecular systems may not generalize well to new systems, limiting their applicability.
- Interpretability: Understanding how deep learning models make predictions is critical, especially in fields like drug discovery where mistakes can be costly.
Despite these challenges, the future of accelerating MD simulation with deep learning is bright. As computational resources grow and deep learning techniques become more refined, the combination of these two powerful tools will continue to revolutionize fields like drug discovery, materials science, and biophysics.
Conclusion
The integration of accelerating MD simulation with deep learning is transforming molecular research by accelerating simulations, improving accuracy, and enabling researchers to explore previously inaccessible timescales and molecular behaviors. From drug discovery to materials science, this combination is opening new frontiers in scientific research, pushing the boundaries of what can be achieved with computational models. As deep learning technologies.
If you want to explore more about applications of accelerating MD Simulation with Deep Learning you can join us in Dubai for an exciting 2.5 Day Masterclass. More information is available HERE