The convergence of artificial intelligence (AI) with molecular dynamics (MD) simulations has opened new horizons in drug discovery. Traditional drug discovery methods are often time-consuming and expensive, but recent advancements in computational tools, particularly applications of MD simulation using Python, have made it possible to explore molecular interactions with greater efficiency and precision. When combined with AI, MD simulations enable researchers to simulate, predict, and optimize drug-target interactions at an atomic level, accelerating the pipeline from hit discovery to clinical development.
In this article, we will explore the key applications of MD simulation using Python in an AI-driven drug discovery pipeline. We will also look at how Python’s flexibility and rich ecosystem of scientific libraries make it a perfect fit for integrating AI into molecular dynamics workflows, facilitating the efficient exploration of drug candidates.
MD Simulations in Drug Discovery: An Overview
Molecular dynamics (MD) simulations model the time-dependent behavior of molecules by applying the laws of classical mechanics. In the context of drug discovery, applications of MD simulation using python allow researchers to:
- Study the dynamic behavior of drug molecules and their targets (proteins, enzymes, receptors) in biological systems.
- Understand molecular interactions, binding affinities, and stability of drug-target complexes.
- Predict the structural and energetic properties of potential drug candidates.
When AI techniques such as machine learning (ML) and deep learning (DL) are incorporated into this process, they enhance the predictive capabilities of MD simulations, making them even more powerful. AI-driven tools can identify patterns, predict molecular behavior, and accelerate key processes such as virtual screening, hit optimization, and lead selection.
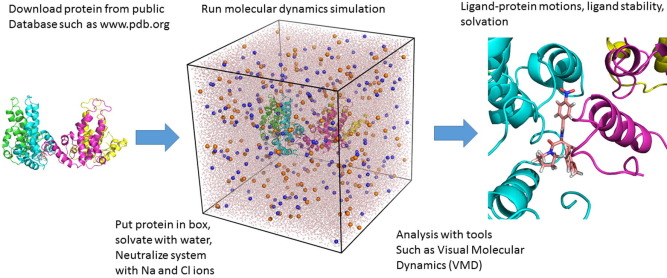
Key Applications of MD Simulation Using Python in AI-Driven Drug Discovery
1. Virtual Screening and Hit Identification
One of the earliest stages in the drug discovery pipeline is identifying potential “hits”—small molecules that exhibit promising interactions with a biological target. Virtual screening involves evaluating a large database of compounds to identify those that may bind to a target protein or receptor.
applications of MD simulation using Python play a key role here by simulating the binding interactions between a drug candidate and its target. Combined with AI models, the accuracy of virtual screening can be significantly improved. AI algorithms can predict the binding affinities and optimize the compounds for better drug-target interactions.
How It Works:
- Molecular Docking: Python-based MD libraries like OpenMM can be used to simulate docking events, where the drug candidate interacts with the binding site of the target protein.
- Machine Learning Models: AI models can learn from these simulations, identifying which molecular features contribute to strong binding affinities. For example, convolutional neural networks (CNNs) can predict binding scores after training on large datasets of docking simulations.
Example Workflow:
- Simulation: Use applications of MD simulation using python to model the drug-target interaction using Python libraries like OpenMM.
- AI Integration: Train an ML model on the simulation data to predict the binding energies and rank the top candidates.
- Refinement: Use reinforcement learning techniques to refine the drug candidates, optimizing molecular structures for improved binding.
By automating the identification of hits applications of MD simulation using python coupled with AI, the virtual screening process becomes faster, more accurate, and capable of handling large compound libraries.
2. Binding Affinity Prediction and Lead Optimization
Once potential hits have been identified, they must be optimized into lead compounds—molecules with higher binding affinities, selectivity, and better pharmacokinetic properties. Applications of MD simulation using python, combined with AI-driven approaches, enable accurate predictions of binding free energies, which is critical for lead optimization.
Binding affinity prediction involves simulating the free energy changes associated with the binding of a drug to its target. Applications of MD simulation using Python tools such as GROMACS and PyEMMA can compute free energy perturbations (FEP) to predict how modifications to a molecule affect its binding strength.
AI-Driven Prediction:
- Deep Learning: AI models, especially deep learning techniques, can be trained to predict binding affinities based on structural and dynamic features of the molecule obtained from MD simulations.
- Transfer Learning: By using transfer learning, AI models can apply knowledge learned from one set of compounds to predict binding affinities for new compounds, saving computational resources.
Example Workflow:
- MD Simulation: Use Python-based tools to simulate the binding interaction and extract dynamic features.
- AI Model Training: Train a deep learning model to predict binding affinities based on the output from MD simulations.
- Lead Optimization: Use AI to optimize molecular properties like solubility, bioavailability, and target selectivity based on predictions of binding affinities.
This AI-enhanced MD simulation approach streamlines the lead optimization phase by providing rapid feedback on potential improvements to molecular structures.
3. Protein-Ligand Interaction Analysis
Understanding how a drug interacts with its target at the atomic level is crucial for drug efficacy and safety. MD simulations are particularly useful for studying protein-ligand interactions over time, providing insights into binding modes, conformational changes, and key interaction residues.
Python libraries such as MDAnalysis and PyMOL allow for detailed analysis and visualization of these interactions, helping researchers identify critical binding sites and potential allosteric sites for drug targeting. AI models can further enhance this process by analyzing large datasets of protein-ligand interactions and identifying patterns that may not be obvious through traditional methods.
Example Applications:
- Conformational Sampling: MD simulations provide multiple snapshots of the protein-ligand complex, capturing conformational changes during binding. AI can be used to classify these conformations and predict which one leads to the strongest binding interaction.
- Interaction Fingerprints: AI can analyze interaction fingerprints—specific patterns of molecular interactions like hydrogen bonds or hydrophobic contacts—to predict the stability and efficacy of drug binding.
Example Workflow:
- Simulation: Run MD simulations using MDAnalysis to model how the ligand binds to the protein and analyze key interaction points.
- AI Analysis: Use AI algorithms to classify binding modes and predict which interactions are most important for strong binding.
By combining MD simulations with AI analysis, researchers can gain deeper insights into protein-ligand interactions and design more effective drugs.
4. Predicting Drug Resistance
One of the major challenges in drug discovery is predicting how mutations in a target protein can lead to drug resistance. MD simulations are highly effective in modeling how mutations affect the structure and dynamics of a protein. AI models trained on these simulations can predict whether a mutation will confer resistance to a drug, enabling the design of more robust drugs that can overcome resistance.
Example Workflow:
- Simulation: Perform MD simulations of the wild-type and mutant forms of a target protein using Python-based tools like OpenMM or GROMACS.
- AI Prediction: Train an AI model on the simulation data to predict the impact of mutations on drug binding.
- Optimization: Use AI-driven design tools to suggest modifications to the drug molecule to counteract resistance.
This application is especially valuable in the context of diseases such as cancer and viral infections, where drug resistance is a significant obstacle to treatment.
5. Drug Toxicity and ADMET Predictions
In addition to efficacy, a drug candidate must also have favorable ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties. Predicting these properties early in the drug discovery pipeline can save time and resources by eliminating candidates with poor pharmacokinetic or toxicological profiles.
By integrating MD simulations using Python with AI-driven models, researchers can predict ADMET properties more accurately. AI models can learn from the dynamic behavior of molecules in simulations to predict properties such as membrane permeability, metabolic stability, and toxicity.
Example Workflow:
- Simulation: Use MD simulations to model the interaction of the drug candidate with biological membranes or metabolic enzymes.
- AI Prediction: Train machine learning models on these simulations to predict ADMET properties.
- Optimization: Use AI algorithms to modify the molecular structure to improve ADMET characteristics.
By incorporating AI into MD simulations, researchers can optimize drug candidates not only for efficacy but also for safety and pharmacokinetic performance.
Python Libraries for MD Simulations and AI Integration
Several Python libraries facilitate the integration of MD simulations and AI models in drug discovery:
- OpenMM: A Python-based toolkit for running MD simulations, offering GPU acceleration for efficient computation.
- MDAnalysis: A Python library for analyzing trajectories and extracting molecular features from simulations.
- PyEMMA: A Python library for constructing Markov state models (MSMs) and analyzing large-scale simulation data.
- scikit-learn: A machine learning library that can be used for training AI models on simulation data.
- TensorFlow/PyTorch: Popular deep learning frameworks for building and training neural networks on molecular data.
Conclusion
The integration of MD simulations using Python with AI-driven methods has transformed the drug discovery process, offering faster, more accurate, and more cost-effective ways to discover and optimize new drugs. By applying Python’s powerful simulation and AI libraries, researchers can simulate molecular interactions, predict binding affinities, study drug resistance, and optimize lead compounds—all within a unified computational framework.
As AI-driven drug discovery continues to advance, the role of MD simulations will only grow in importance, enabling more personalized and precise treatments for a wide range of diseases.
If you want to explore more about applications of Running MD Simulation using Python you can join us in Bengaluru for an exciting 1 Day Training. More information is available HERE