The rapid evolution of computational tools has transformed the pharmaceutical industry, particularly in the field of computer-aided drug discovery (CADD). Traditional drug discovery processes, often lengthy and expensive, have benefited immensely from automation, data science, and machine learning technologies. Python, with its extensive suite of scientific libraries and tools, plays a pivotal role in automating drug discovery pipelines, enabling researchers to accelerate the journey from raw data to potential drug candidates. This article explores how Python automates various stages of the drug discovery process, from handling biological data to generating new drug candidates.
The process of discovering new drugs has always been long, expensive, and fraught with challenges. However, the advent of computer-aided drug discovery (CADD) has revolutionized this field by incorporating computational tools to streamline various stages of drug development. One of the key enablers of this transformation is Python, a powerful, open-source programming language that has been widely adopted by the scientific community which acts as a catalyst for automating drug discovery pipelines. With its rich ecosystem of libraries and frameworks, Python is helping researchers automate entire drug discovery pipelines, accelerating the process from data collection to identifying promising drug candidates.
In this article, we’ll explore how Python can be used to automate different steps in the computer-aided drug discoverypipeline and its impact on the efficiency and accuracy of modern drug discovery.
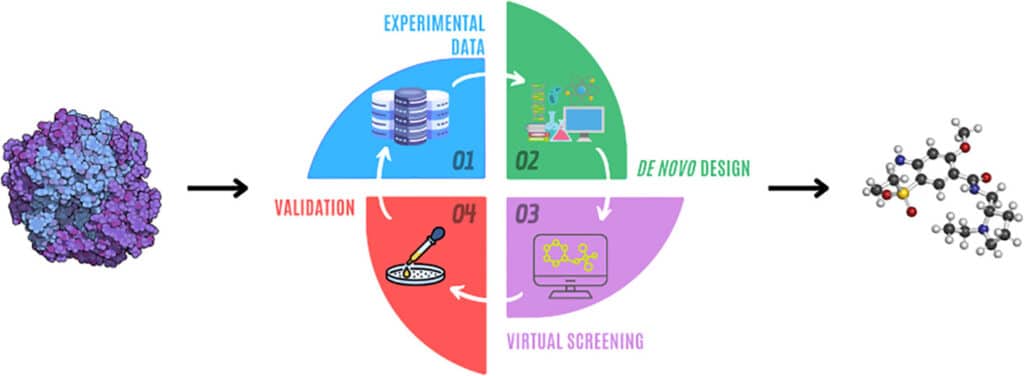
What is Computer-Aided Drug Discovery (CADD)?
Computer-aided drug discovery (CADD) involves the use of computational techniques to identify and optimize potential therapeutic compounds. The drug discovery process typically spans multiple stages:
- Target Identification and Validation: Identifying biological targets (e.g., proteins or enzymes) involved in diseases.
- Hit Discovery: Screening large libraries of compounds to find potential “hits” that bind to the target.
- Lead Optimization: Refining the hit compounds to improve their efficacy, specificity, and safety.
- Preclinical Testing: Evaluating drug candidates in biological systems.
Automation plays a crucial role in optimizing each of these steps, reducing the need for costly and time-consuming laboratory experiments. Python, with its powerful libraries and flexibility, has emerged as a key tool for automating the drug discovery pipeline, bringing the power of data analysis, molecular modeling, and machine learning to researchers’ fingertips.
Why Python for Automating Drug Discovery Pipelines?
Python’s popularity in the scientific community stems from its simplicity, versatility, and a vast array of libraries tailored for data analysis, molecular modeling, and machine learning. Here are a few reasons why Python is the go-to language for automating drug discovery pipelines:
- Ease of Use: Python’s clear syntax makes it easy to learn, use, and deploy in real-world research settings.
- Rich Library Support: Libraries such as RDKit, PyMOL, Open Babel, and Scikit-learn offer ready-to-use tools for cheminformatics, molecular visualization, and machine learning.
- Interoperability: Python can be integrated with established computational chemistry software like AutoDock, GROMACS, and OpenMM.
- Automation: Python scripts can automate tedious tasks like screening compounds, setting up simulations, and analyzing results, enabling researchers to focus on critical decision-making.
Now, let’s break down how Python is used to automate each phase of the computer-aided drug discovery pipeline.
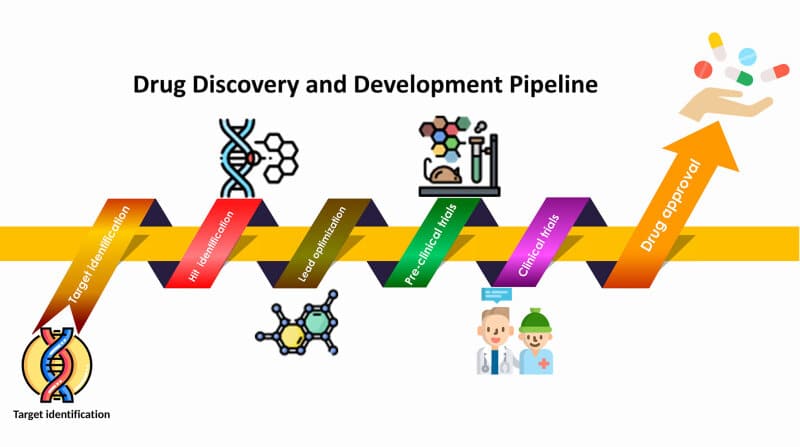
Key Steps in Automating Drug Discovery Pipelines with Python
1. Data Collection and Preprocessing
The first step in any drug discovery project is gathering and preprocessing large datasets, which may include information on biological targets, chemical compounds, and molecular interactions. Python offers robust tools for handling both structured and unstructured data from sources such as PubChem, ChEMBL, and Protein Data Bank (PDB).
- Pandas and NumPy: For handling large datasets, these libraries allow researchers to clean, filter, and manipulate data.
- RDKit: A popular cheminformatics library for working with chemical structures, RDKit is ideal for converting data formats (e.g., SMILES, SDF) and generating molecular descriptors.
Python automates the preprocessing of large compound datasets, ensuring that only drug-like molecules move forward in the pipeline.
2. Virtual Screening and Molecular Docking
One of the most critical stages in computer-aided drug discovery is virtual screening, which involves screening large compound libraries to identify potential drug candidates. Virtual screening can be automated using Python to rank and select compounds based on their binding affinity to a target protein.
Python’s AutoDockTools and PyRx allow researchers to set up, run, and analyze molecular docking experiments using tools like AutoDock Vina.
By automating molecular docking using Python, researchers can quickly screen thousands of compounds, enabling faster identification of promising hits for further testing.
3. Lead Optimization with Machine Learning
After identifying hits through virtual screening, the next step is lead optimization, where the drug candidates’ chemical properties are refined to enhance their efficacy and reduce toxicity. Machine learning (ML) has become a powerful tool for optimizing drug candidates by predicting the biological activity of compounds based on their chemical structure.
Python’s scikit-learn, TensorFlow, and PyTorch libraries provide the foundation for building ML models that can predict important properties such as bioavailability, toxicity, and binding affinity.
By integrating machine learning models, Python can automate the optimization of drug candidates, allowing researchers to focus on compounds with the highest potential for success.
4. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations allow researchers to study the behavior of drug candidates at an atomic level over time, providing insights into their stability, binding mechanisms, and interaction with biological targets. Python’s OpenMM and MDAnalysis libraries make it easy to automate MD simulations, allowing researchers to simulate complex molecular systems and analyze the results.
MD simulations provide insights into the dynamic behavior of drug candidates, helping researchers optimize their binding affinity and overall stability.
5. QSAR Modeling for Predicting Drug Activity
Quantitative Structure-Activity Relationship (QSAR) models use machine learning to predict the biological activity of compounds based on their chemical structure. Python’s RDKit and scikit-learn libraries are widely used to build QSAR models, which allow researchers to screen large libraries of compounds for potential drug candidates.
QSAR modeling is a vital part of computer-aided drug discovery, allowing researchers to predict the activity of new compounds based on their molecular features.
6. Automating the Workflow with Python
Python’s flexibility allows researchers to integrate various steps of the drug discovery pipeline into an automated workflow. By combining data preprocessing, virtual screening, machine learning, molecular dynamics simulations, and QSAR modeling into a single pipeline, Python automates the discovery and optimization of drug candidates.
Automating drug discovery pipelines allows researchers to focus on analyzing the results and making critical decisions, while Python handles the repetitive and computationally intensive tasks.

Conclusion
The integration of Python into computer-aided drug discovery has transformed the drug discovery pipeline, enabling automation at every step. From data collection and virtual screening to molecular dynamics simulations and machine learning-based lead optimization, Python’s powerful libraries and flexible frameworks allow researchers to streamline workflows and accelerate the development of new therapeutics.
By automating these processes, researchers can significantly reduce the time and cost associated with traditional drug discovery, increasing the likelihood of identifying
If you want to explore more about applications of Introduction to Computer Aided Drug Discovery with Python you can join us in Bhopal for an exciting 2.5 Day Masterclass. More information is available HERE