Molecular dynamics (MD) simulations have long been a cornerstone in the study of molecular systems, providing detailed insights into the behavior of atoms and molecules over time. These simulations allow scientists to observe the interactions between molecules at an atomic scale, making them crucial in areas such as drug discovery, material science, and biophysics. However, despite their invaluable contributions, traditional MD simulations are computationally expensive and time-consuming, especially for complex biological systems or large-scale simulations.
Enter deep learning—an advanced subset of artificial intelligence (AI)—which has opened new frontiers in MD simulations by offering faster, more accurate predictions and the ability to model large systems efficiently. The integration of MD simulation with deep learning is revolutionizing computational chemistry, enabling researchers to accelerate simulations, improve accuracy, and explore molecular behaviors that were previously inaccessible.
In this article, we will explore how deep learning is transforming MD simulations, the challenges it addresses, and the tools and frameworks that make this integration possible.
What Are MD Simulations?
Molecular dynamics (MD) simulations involve calculating the motion of atoms and molecules over time using Newton’s laws of motion. These simulations are widely used in computational biology and chemistry to predict the physical movements of molecular systems such as proteins, DNA, and drug molecules. MD simulations offer a time-resolved picture of how molecules behave, how they fold, and how they interact with each other.
Key Applications of MD Simulations:
- Protein Folding: Understanding how proteins adopt their functional shapes.
- Drug-Protein Interactions: Studying how drug molecules bind to their biological targets.
- Materials Science: Investigating the properties of materials at the atomic level.
However, one of the biggest limitations of MD simulations is their computational cost. Accurate MD simulations require solving millions of equations of motion, which can take days or even weeks to complete, particularly when studying large biological systems or simulating long timescales.
Limitations of Traditional MD Simulation:
- Computationally Intensive: High-resolution simulations require immense computational resources.
- Limited Timescales: MD simulations are often restricted to short timescales (nanoseconds to microseconds), while many biological processes occur over milliseconds or longer.
- Scaling Issues: Simulating large molecular systems or interactions involving thousands of atoms can become prohibitively expensive.
This is where deep learning steps in to revolutionize the process.
The Role of Deep Learning in MD Simulation
Deep learning involves the use of neural networks—algorithms that mimic the human brain’s structure—to identify patterns and make predictions. By training on large datasets, these models can learn complex relationships between inputs and outputs. In the context of MD simulations, deep learning can be used to:
- Accelerate Simulations: Deep learning models can predict the future states of molecular systems without the need to compute every atomic interaction step-by-step, speeding up simulations significantly.
- Generate More Accurate Models: Deep learning can refine force fields (the set of rules that govern molecular interactions) to improve the accuracy of simulations.
- Reduce Computational Costs: By learning from previous simulations, deep learning models
Accelerating MD Simulation with Deep Learning
One of the most significant contributions of deep learning to MD simulations is its ability to accelerate the process without sacrificing accuracy. Traditional MD simulations require calculating forces at every time step, which is computationally expensive. However, deep learning models can “learn” the dynamics of molecular systems and make predictions about future states without needing to calculate every force interaction.
Example: Time-Lagged Autoencoders (TAEs)
Time-Lagged Autoencoders (TAEs) are a type of neural network used to accelerate MD simulations. By training on time-lagged data from simulations, TAEs learn to predict the future configuration of a molecular system based on its past states. This allows researchers to skip computationally expensive steps, accelerating the simulation process while maintaining accuracy.
Enhancing Force Fields with Deep Learning
Force fields, which describe how atoms interact with each other, are fundamental to MD simulations. Traditional force fields are often based on simplified approximations of molecular interactions, which can limit their accuracy. Deep learning models can be used to develop more accurate, data-driven force fields by training on high-resolution quantum mechanical calculations and experimental data.
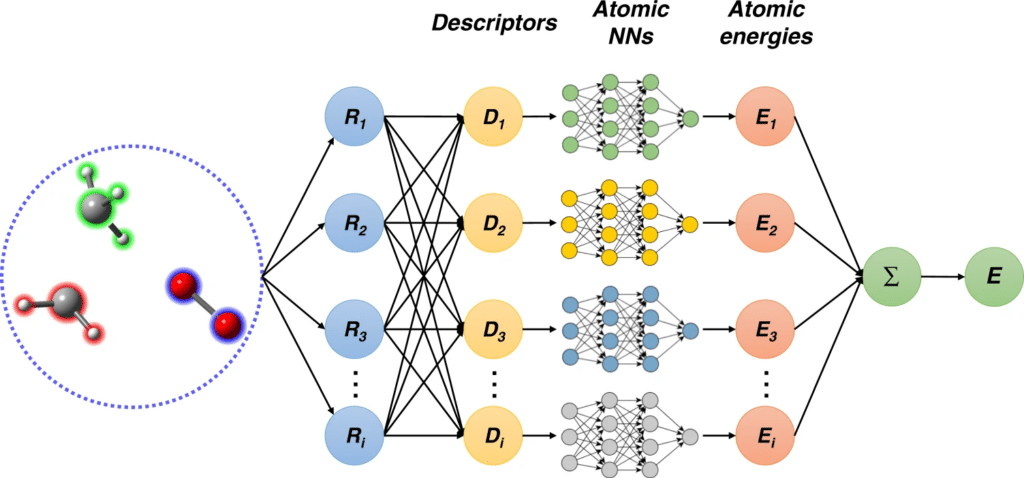
Example: Neural Network Potentials (NNPs)
Neural network potentials (NNPs) are deep learning models trained to predict atomic interactions more accurately than traditional force fields. By learning from quantum mechanical simulations, NNPs can model complex molecular systems with greater precision, capturing subtleties such as electronic effects that traditional methods may overlook.
Predicting Long Timescale Dynamics
One of the biggest challenges in MD simulation with Deep Learning is capturing long timescale dynamics, such as protein folding or conformational changes, which often take place over milliseconds or even seconds. These processes are crucial for understanding biological function but are difficult to simulate using traditional MD methods.
Deep learning models offer a solution by learning from short timescale data and predicting how molecular systems behave over longer periods. This allows researchers to simulate processes that were previously inaccessible using traditional methods.
Example: Markov State Models (MSMs)
Markov state models (MSMs), combined with deep learning, have become a powerful tool for predicting long timescale dynamics. MSMs break down the dynamics of a molecular system into a series of states, and deep learning models are used to predict transitions between these states over long timescales. This allows researchers to simulate complex molecular behaviors, such as protein folding pathways, in a fraction of the time required by traditional MD simulations.
Key Tools for Integrating MD Simulation with Deep Learning
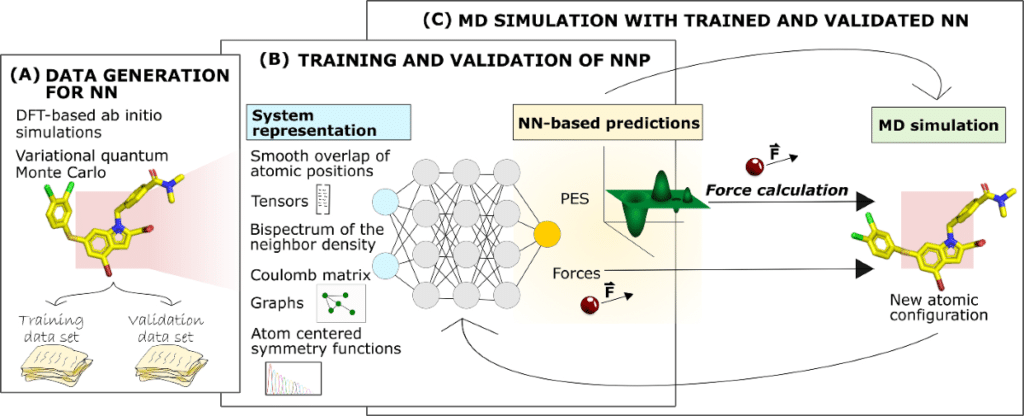
The integration of MD simulation with deep learning is made possible by a variety of Python-based libraries and frameworks that enable researchers to build and train neural networks for molecular simulations.
1. TensorFlow and PyTorch
Both TensorFlow and PyTorch are popular deep learning libraries that are widely used in the scientific community. These libraries provide tools for building and training neural networks and have been instrumental in developing MD Simulation with Deep Learning.
- TensorFlow: TensorFlow is a scalable machine learning framework that offers robust support for both neural network training and inference, making it ideal for large-scale simulations.
- PyTorch: Known for its flexibility and ease of use, PyTorch is popular among researchers for developing custom models and prototyping new algorithms.
2. OpenMM
OpenMM is an open-source toolkit that enables the simulation of molecular systems and integrates seamlessly with Python. Researchers can use OpenMM to set up MD simulations with Deep Learning and then apply deep learning models to accelerate or enhance the results. OpenMM provides flexibility in defining force fields and handling large biomolecular systems.
3. DeepDriveMD
DeepDriveMD is a framework designed for coupling MD simulations with deep learning models, enabling large-scale simulations and training. It provides tools for running simulations on high-performance computing (HPC) clusters and using deep learning to steer the simulation based on real-time data.
4. MDAnalysis
MDAnalysis is a Python package for analyzing MD simulations. It allows researchers to process large amounts of simulation data and apply machine learning techniques to extract insights. Deep learning models can be integrated with MDAnalysis to automatically classify molecular states or predict future configurations.
Challenges and Future Directions
While the integration of MD simulation with deep learning has shown tremendous promise, several challenges remain. These include:
- Data Requirements: Deep learning models require large datasets for training, and generating accurate training data from quantum mechanical calculations can be computationally expensive.
- Model Interpretability: Deep learning models can be challenging to interpret, making it difficult to understand how predictions are being made and whether they are physically meaningful.
- Scalability: Scaling deep learning-enhanced MD simulations to very large systems or extremely long timescales is still an area of active research.
Despite these challenges, the future of MD simulation with deep learning looks bright. Advances in AI, coupled with increasing computational power, will continue to push the boundaries of what can be simulated and predicted. The integration of deep learning into MD simulations has the potential to unlock new discoveries in drug design, materials science, and molecular biology.
Conclusion
The integration of MD simulation with deep learning represents a new frontier in computational chemistry. By leveraging the power of deep learning, researchers can accelerate simulations, improve the accuracy of force fields, and explore molecular dynamics on previously inaccessible timescales. As deep learning models become more sophisticated and computational resources continue to grow, the combination of MD simulations and AI will undoubtedly play a central role in shaping the future of molecular discovery.
If you want to explore more about applications of Integrating MD Simulation with Deep Learning you can join us in Dubai for an exciting 2.5 Day Masterclass. More information is available HERE