The pharmaceutical industry is continuously evolving, with the demand for faster and more cost-effective drug discovery growing every day. Traditionally, drug discovery was a long, expensive process, taking years of trial and error before a successful drug could be developed. However, with the introduction to computer aided drug discovery (CADD), researchers now have powerful computational tools at their disposal that can streamline this process, reduce costs, and improve the accuracy of drug development. In this article, we will explore how computer-aided drug discovery works and how Python has become a vital tool for researchers entering this field.

What is Computer-Aided Drug Discovery?
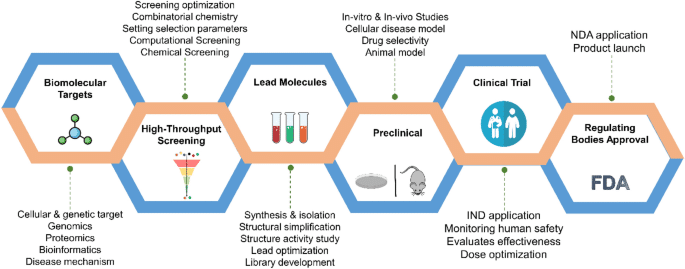
Computer-aided drug discovery (CADD) refers to the use of computational techniques and tools to assist in the identification and development of potential therapeutic compounds. These methods include molecular docking, virtual screening, molecular dynamics simulations, and quantitative structure-activity relationship (QSAR) modeling, among others.
CADD enables researchers to model the behavior of biological systems and predict how drug-like molecules will interact with biological targets such as proteins, enzymes, and receptors. This reduces the need for expensive and time-consuming laboratory experiments in the early stages of drug development.
There are two main types of CADD approaches:
- Structure-based drug discovery (SBDD): This approach uses the 3D structure of a biological target (usually a protein) to design and optimize drug candidates.
- Ligand-based drug discovery (LBDD): This approach relies on the knowledge of known active compounds (ligands) to predict the activity of new compounds.
Why Python for Introduction to Computer Aided Drug Discovery?
Python is a preferred language in the scientific community due to its ease of use, extensive libraries, and strong support for computational chemistry and biology applications. Python offers several advantages of Introduction to computer-aided drug discovery:
- Accessibility: Python’s simple syntax makes it easy for beginners to learn and apply.
- Powerful Libraries: Python has a rich ecosystem of libraries for molecular modeling, data analysis, machine learning, and visualization, making it a versatile tool for CADD.
- Interoperability: Python can interface with other tools and software commonly used in CADD, such as AutoDock, GROMACS, and OpenMM, allowing for seamless integration into existing workflows.
This makes Python an ideal choice for both beginners and experienced researchers looking to apply computational methods to drug discovery.
Key Applications of Computer-Aided Drug Discovery Using Python

1. Molecular Docking
One of the core components of Introduction to computer aided drug discovery is molecular docking. Docking simulations aim to predict the preferred orientation of a small molecule (drug candidate) when bound to its target protein. This helps in identifying how well the drug binds and its potential therapeutic efficacy.
Python can be used to automate and run molecular docking simulations using popular docking software like AutoDockor Open Babel. For example, PyAutoDock provides a Python interface for running AutoDock simulations, making it easier to set up and analyze docking experiments.
Example Workflow:
- Set Up the Docking Simulation: Use Python scripts to prepare the target protein and ligand structures, often starting with a PDB file for the protein and a mol2 or SDF file for the ligand.
- Run Docking: Run the docking simulations using AutoDock from within Python.
- Analyze Results: Use Python-based libraries such as MDAnalysis or PyMOL to visualize the docking poses and calculate binding energies.
2. Virtual Screening
Virtual screening is another critical application of CADD, where large compound libraries are computationally screened to identify those most likely to bind to a biological target. Python allows you to automate this process and evaluate thousands of compounds in a fraction of the time compared to traditional methods.
Python libraries such as RDKit and Open Babel are commonly used to manipulate chemical structures and run virtual screening workflows. These libraries allow you to:
- Filter compounds based on molecular properties such as molecular weight, solubility, and toxicity.
- Calculate molecular fingerprints and similarity scores.
- Perform high-throughput virtual screening using molecular docking or machine learning models.
Example Workflow:
- Generate a Compound Library: Use RDKit to import, filter, and manipulate chemical structures.
- Docking: Run molecular docking for each compound in the library using PyAutoDock or a similar tool.
- Rank the compounds based on their docking scores or binding affinities.
3. Molecular Dynamics (MD) Simulations
MD simulations are widely used in the Introduction to computer aided drug discovery to study the dynamic behavior of drug molecules and their interactions with biological targets over time. Python libraries such as OpenMM and MDAnalysis allow users to set up and analyze MD simulations to explore:
- Protein-ligand interactions.
- Conformational changes in the target protein.
- Binding affinities over time.
Example Workflow:
- Prepare the System: Use Python-based tools like OpenMM to set up the protein-ligand system, including defining force fields and initial conditions.
- Run the Simulation: Use GPU-accelerated MD simulations to observe the dynamic behavior of the complex.
- Analyze the Results: Extract key information such as root mean square deviation (RMSD), hydrogen bonds, and binding energies using MDAnalysis.
4. QSAR Modeling and Machine Learning
Quantitative Structure-Activity Relationship (QSAR) modeling is an essential technique in drug discovery that uses chemical structures to predict biological activity. Machine learning models can be trained on chemical datasets to build QSAR models that predict the efficacy and toxicity of new compounds.
Python libraries like scikit-learn, RDKit, and XGBoost are commonly used to build machine learning models for QSAR analysis. By combining chemical descriptors (e.g., molecular fingerprints) with predictive models, researchers can quickly evaluate the potential of new drug candidates.
Example Workflow:
- Feature Extraction: Use RDKit to calculate molecular descriptors or fingerprints for each compound.
- Model Training: Train a machine learning model using scikit-learn or XGBoost to predict biological activity.
- Prediction: Use the trained model to predict the activity of new compounds.
If you want to explore more about applications of Introduction to Computer Aided Drug Discovery with Python you can join us in Bhopal for an exciting 2.5 Day Masterclass. More information is available HERE