The most important part of a drug discovery process after getting the interaction is checking the stability of that interaction. As during the process of Molecular Docking, we are looking for interaction between our Protein of Interest & the Ligand molecule with therapeutic effect. Now if we are able to get the interaction between them then the most important question arises whether the interaction between the two is stable or not.
Hence, we need to perform the process of Molecular Dynamics Simulation which helps us in understanding the stability of the protein-ligand complex molecule. This is also one of the major applications of Bioinformatics. If you want to learn more about the application of Bioinformatics you can learn HERE. So, lets first dive into molecular dynamics simulation basics and understand how it works.
What is Molecular Dynamics Simulation?
Molecular Dynamics Simulation is a computer program designed to calculate the stability of a macro-molecule be it a Protein, Protein-Ligand Complex, Protein-DNA Complex, Protein-Protein Complex Etc.
With Molecular Dynamics Simulation we are examining the tangible movements of atoms and molecules. In the most standard version, the orbits of atoms and molecules depends on arithmetically solving Newton’s equations of motion for a system of collaborating particles, where forces between the particles and their potential energies are enumerated using interatomic potentials or molecular mechanics force fields.
We deliver MD Simulation services for different types of complexes like Protein-Ligand Complex, Protein-Protein Complex, Enzyme-Substrate Complex, DNA -Protein Complex. The scanning of result encompasses various graphs like RMSD, ROG Etc.
Molecular Dynamics Simulations, as a significant supplement to conceptual calculations and experimental methods as well as a beneficial technique to productively understand macromolecular structure-to-function correlation. It has unfolded into a mature methodology frequently seen in the province of chemistry, biomedicine and physics.
As molecular systems thereabouts consist of an ample number of particles, it is unobtainable to determine the properties of these composite systems analytically; MD simulation overcomes this problem by using exponential methods. However, long MD simulations are often analytically ill-conditioned, generating mounting mistakes in numerical incorporation that can be slashed with suitable selection of algorithms and parameters, but not dropped entirely.
We take the lead of the Molecular Dynamics simulation mechanism to assist customers foresee the time dependent switches in a protein system, which can accommodate proteins, DNAs/RNAs, lipids and other minor ligands, allowing for the consideration of events of biological and pharmaceutical significance.
Specifically, simulation can be implemented to indicate protein flexibility, refine experimentally determined structures, assess protein-ligand binding, test bio-catalysis or even perceive the protein folding process.Molecular Dynamics Simulation can be used to explore conformational space, and is repeatedly the method of choice for large molecules such as proteins. In this, the energy facet is inspected by solving Newton’s laws of motion for the system.
There are so many distinct models, brands, and recasting of hardware that selecting the precise components for your system can be an immense challenge. Determination of what stipulations impact molecular dynamics modelling and simulations the most, has been done already.
STEPS Involved in Running the Molecular Dynamics Simulation
There are a number of steps involved in running the process of Molecular Dynamics Simulation which the exact same steps involved in Molecular Dynamics Simulation tutorial also are as follows: Taking the case of running Molecular Dynamics Simulation of Protein-Ligand Complex
The first step in every Molecular Dynamics Simulation process is Generating the topology of our Macro-Molecule which in majority of cases is our Protein or Enzyme.
There are a number of options available for choosing the forcefield for generating your Topologies. As the forcefields are a very major component of topology. Some of the examples of available forcefields are: OPLS-AA, AMBER, GROMOS & CHARMM. So, we can choose any one of them for generating the topology of our Macro-Molecule.
**But we need to remember that which ever forcefield we are using to generate the topology of our Macro-Molecule same forcefield we need to choose for our other Micro or Macro-Molecule. Otherwise, your process will end-up with so many errors.
The Second Step involves generating the topology of your Micro-Molecule which in most of the cases is a chemical compound which we are going to use as a lead molecule.
The third Step involves creating the complex back again as we had broken up the complex in the beginning for generating the topologies.
Note: Even though we are breaking the complex and making it back again does not mean that we can skip the process of Molecular Docking and directly jump to Molecular Dynamics Simulation If you do so the process will be wrong as we need to perform the process of Molecular Docking in order to check whether the interaction between the protein and Ligand is there or not. If it’s there in that case only we can perform Molecular Dynamics Simulation on that Molecule.
Then we need to include the topology information of our ligand molecule (micro-molecule) in the main topology so that the software is able to identify co-ordinates of our ligand in the complex and continue the process.
The fourth step involves defining a box around our complex which can fit the whole complex inside it and filling the box with the solvent (water) molecules.
The fifth step involves adding ions to our complex so that it can be neutralized by adding the required amount of NA+ or CL– ions. Once the ions have been added now our system is ready for further processing.
The sixth step involves minimizing the energy of our system so that it is stable enough to undergo the process of MD Simulation.
The seventh step involves creating the index for all the components of our system as well as creating the separate Index for our ligand also. As everything has been setup for the process now it’s time for equilibrating our system and performing the main MD run.
The eighth step involves equilibrating our system for NVT & NPT runs which involves equilibrating our system with Volume & Temperature and Pressure & Temperature and then finally it’s time to initiate the final Molecular Dynamics Run which may last for approximately 12-18 hours on a good regular desktop computer system. The time taken completely depends upon the capacity of your system to run the process.
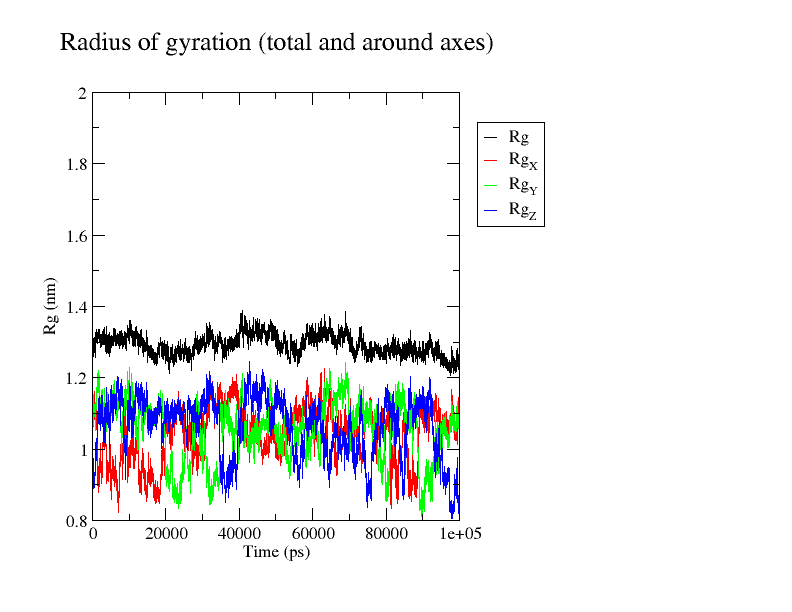
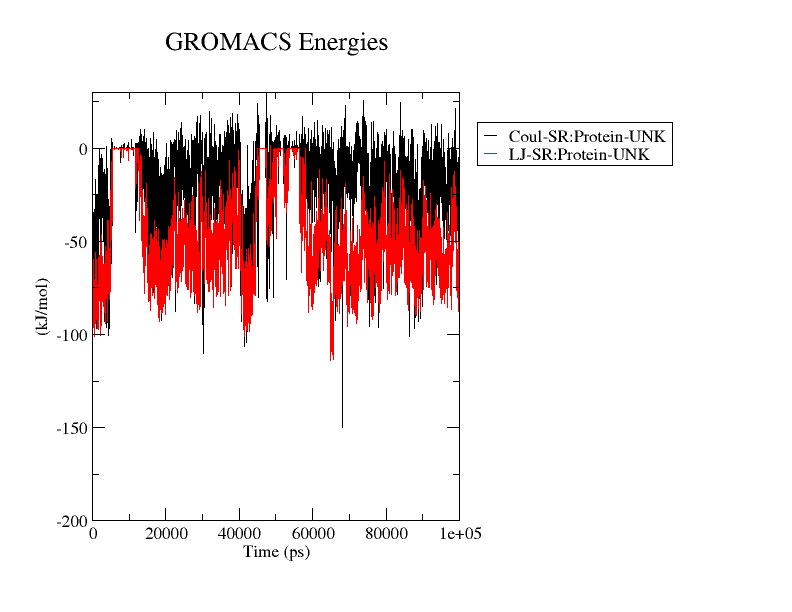
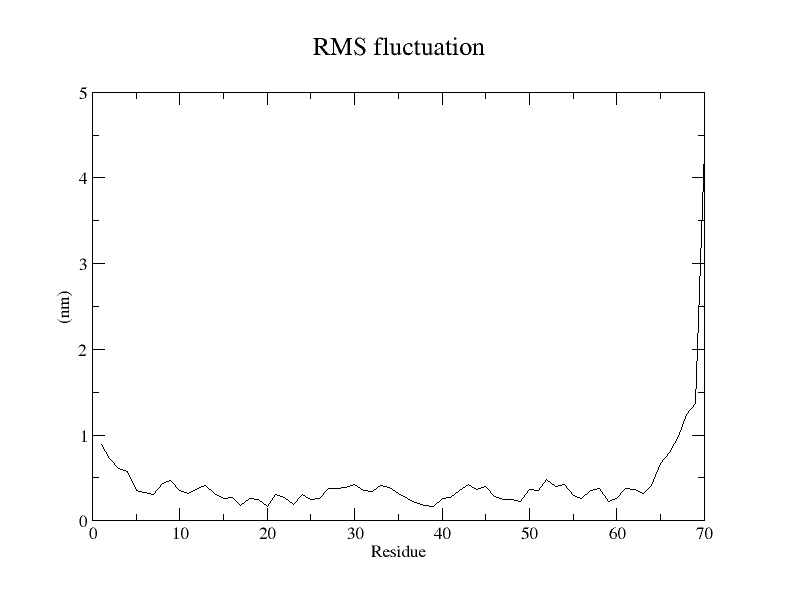
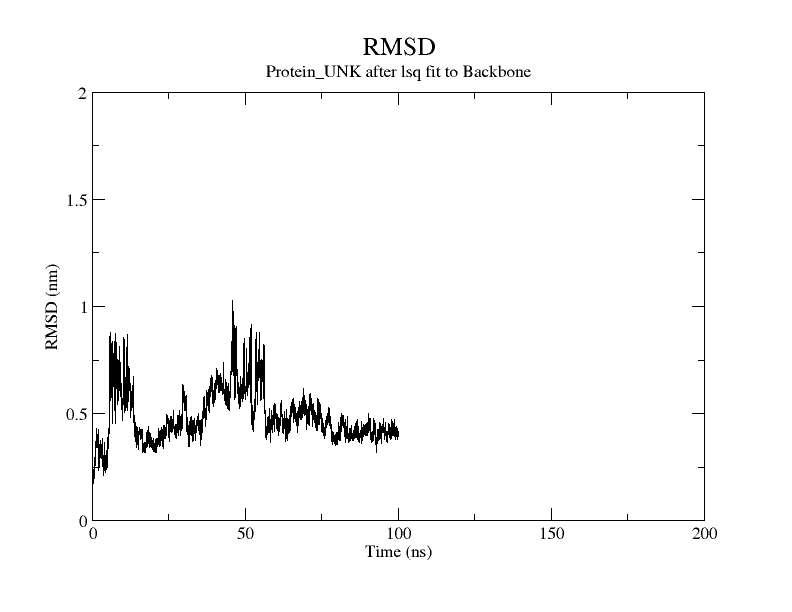
The ninth step involves result interpretation for the whole process by generating a number of graphs like Radius of Gyration (ROG), Root Mean Square Fluctuation (RMSF), Root Mean Square Deviation (RMSD), Interaction Energy (IE), Solvent Accessible Surface Areas (SASA) & Principal Component Analysis (PCA).
We can go about all these graphs and identify whether the results generated from the main Molecular Dynamics Simulation Run are good or not. Further if we need to do the advance analysis one can opt for advance analysis by performing MMPBSA analysis which we will talk about in a separate article.
 |  |
 |  |
Software tools for Running Molecular Dynamics Simulation
There are number of tools available for running Molecular Dynamics Simulation like VMD/NAMD, Desmond, Amber, GROMACS Etc. You can choose any one of these tools based on the comfort of your usage. Like VMD/NAMD can be run on a Windows PC itself you don’t need to install Ubuntu OS for it. Whereas other software like Desmond, Amber & GROMACS needs Ubuntu OS for running the process.
Desmond being a very straight forward tool gets your job done quickly. Amber is a bit tricky one but you can run the process very easily with it once you understand how to do the process with it. GROMACS is a bit tough to install & operate tool but GROMACS is considered as thumb rule for Molecular Dynamics Simulation.
People have a lot of myths around running GROMACS simulation that you need to know coding & programming to run GROMACS but that is not a case anyone who knows how to operate a computer systems and little common sense can easily use the GROMACS software.
Now, you may have one more doubt that why we need Ubuntu OS for running all theses software tools? So, a simple answer to that is Windows OS is a pretty heavy operating System with a lot of graphics & functionality and Molecular Dynamics Simulation is also a very heavy process to run so these two heavy computations will run parallel there a lot of chances that your computer system may crash or even worse burn down.
Hence, we opt for a lighter operating system which has functionality with now much graphics. That’s why we use Ubuntu for running the process. So, the whole computational power can be dedicated to running our process.
Luckily, these programs scale almost straight away so there is an unmediated correlation between the escalation in cores and/or GPUs to the increase in performance that you will get in return. Normally, the most analytically high-priced segment of a MD simulation is the assessment of these long-range electrostatic interplay.
In order to conclude I would like to say that first focus on your pipeline for performing the Molecular Docking process once that is done right you can process for Molecular Dynamics Simulation to understand the stability of your molecules for better results and discovery of good lead molecules.
To know more or to learn how to run this process on your system you can enroll for our 3 Hour Short Course on Drug Discovery, you can register HERE
A brief introduction guide on Molecular Docking & MD Simulation can be accessed HERE for better understanding of the whole process.