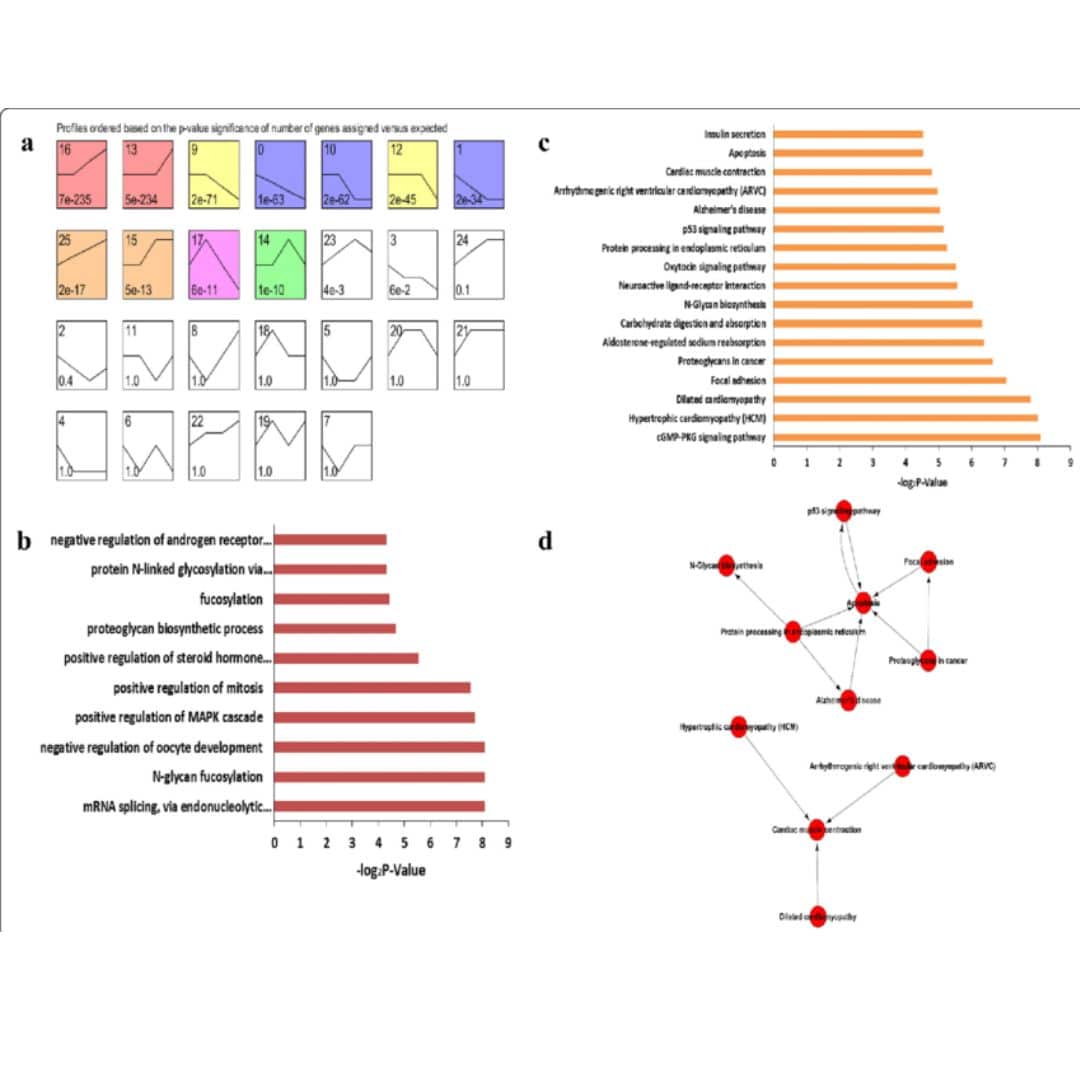
Python for variant calling with VCF file is becoming an essential approach in genomic research, offering a robust and flexible solution for analyzing genetic variants. Variant calling is the process of identifying differences in DNA sequences, such as single nucleotide polymorphisms (SNPs) and insertions or deletions (indels), from sequencing data. The resulting data is often stored in Variant Call Format (VCF) files, a standard file format that contains information about genetic variants and their associated metadata.
Python, with its extensive libraries and tools, provides a streamlined workflow for variant calling and subsequent analysis of VCF files. Libraries like PyVCF, Pandas, and Scikit-allel allow researchers to efficiently parse, filter, and manipulate VCF data, making it easier to identify meaningful genetic variants and interpret their biological significance.
One of the significant advantages of using Python for variant calling with VCF file is its ability to handle large genomic datasets. Python’s data manipulation capabilities enable researchers to filter out low-quality variants, annotate genetic variants with additional information, and perform statistical analyses to identify variants associated with specific traits or diseases.
Applications of Python for variant calling with VCF file
In this section we are discussing some of the potential areas of application of Python for variant calling with VCF file & how it can be helpful in these areas to carryout analysis.
In population genomics, Python is used to analyze genetic variants across different populations to study genetic diversity, population structure, and evolutionary history.
Python facilitates genome-wide association studies (GWAS) by allowing researchers to analyze variants from VCF files in relation to disease phenotypes.
Python can be used to analyze an individual’s VCF file to identify variants that might influence drug response, risk of developing certain diseases, or other personalized health outcomes.
Python enables the prediction of the functional impact of genetic variants identified in VCF files.
Different Types of Analysis
Majorly, there are 3 different types of analysis using Python for variant calling with VCF file so below we have tried to list them down to help you take better decision if this masterclass will be relevant for your learning and be helpful in your intended area of research.
QC & Filtering Analysis
Quality control is essential in variant calling to ensure that only high-confidence variants are retained for further analysis.
Variant Annotation Analysis
Once variants are identified, Python can be used to annotate them with additional biological information, such as gene names.
Allele Frequency Analysis
Python is often used to analyze VCF files in the context of population genetics. Researchers can calculate allele frequencies.
Detailed Agenda of The Workshop
Python’s integration with other bioinformatics tools and frameworks allows for seamless automation of variant calling pipelines. Researchers can write custom scripts to automate repetitive tasks, such as quality control, variant filtering, and annotation, significantly reducing the time and effort required for genomic analysis.
Python for variant calling with VCF file is a powerful and versatile tool, enabling researchers to conduct comprehensive genomic analyses with precision and efficiency, ultimately advancing our understanding of genetics and improving healthcare outcomes.
Apart from the topics mentioned above there are a few extra things which you can take away from this workshop, which will be adding more value to your work
Introductory Documentation
An introductory theory document to help you better understand the subject will also be provided.
Trainers Slide Deck
After the completion of the session complete access to the trainers slide deck will also be provided
Access to Trainers Repo
We will also be providing a complete access to trainers repository so that you can use it as reference later
8+ Hours of Live Training
During the course of 3 days we will be have live8+ hourse of training sessions with the participants.
Guided Hands-On Exercises
Hands-On exercises are a must to better learn any technology and be able to reproduce it later.
Participation Certificate
A Participations certificate is a must after successfully completing the training as a sign of accomplishment.
Expected Outcomes of the Workshop
After completing all the tasks of this workshop all of our participants will be able to:
High-Confidence Genetic Variants
By using Python for variant calling and applying rigorous filtering criteria, researchers can identify high-confidence genetic variants.
Population-Specific Insights
Through the analysis of allele frequencies and variant distribution across different populations, Python can reveal population-specific genetic variants.
Annotation of Genetic Variants
Python enables the automation of variant annotation processes, providing detailed information about each variant's location, associated
Disease Association Studies
Python allows for the integration of variant data with phenotypic information, enabling researchers to conduct GWAS and identify variants
All fee paid is not refundable so please read all the terms & conditions before making any payments. If you still have any doubts please contact us and confirm and then only make the payment.
Participants need to bring their registration tickets along with a valid Institutional ID, then only they will be allowed to attend the session. Please reach out to our team in case of any exceptions.
Please fill all your details in the form correctly as those details will be used in your certificate as well.
Participants need to bring their own computer (laptop) system for the program.
The software tools and other required software tools will be provided from our side for the purpose of this program.
Participants need to reach the venue and report 30 minutes prior to the start of the sessions.
Participants need to wear masks all the time inside the premises and abide by the other rules at the premises.
Particpants need to attend all the sessions in order to be eligible for getting the certificate.
Welcome email will be sent to all the participants with all the details related to the program. Please check your Inbox/Spam folder for the email.
All the details of the software installations and how to prepare your system for the Program will be shared with all the participants in the Welcome Email itself.
Contact Us
We understand that you may have some questions before you make the payment for the course. Feel free to get in contact with us through the below given options.