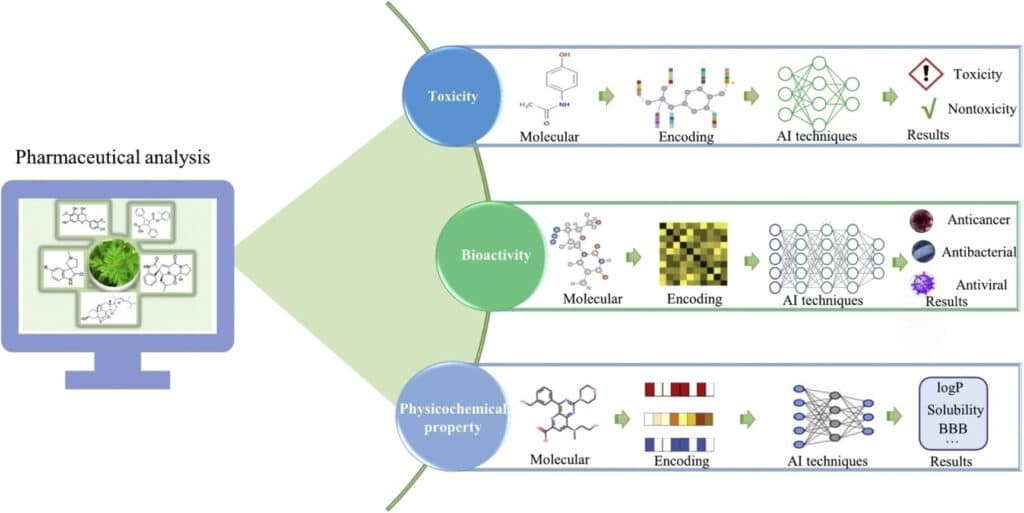
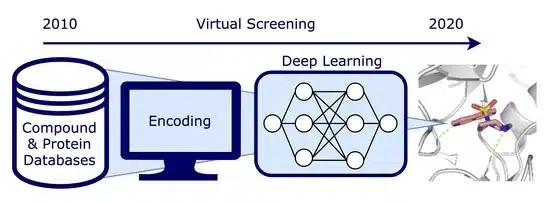
In the world of computer-aided drug discovery (CADD), virtual screening has become an indispensable tool. This technique enables researchers to sift through vast libraries of chemical compounds to identify those most likely to interact with biological targets, significantly reducing the time and cost associated with traditional drug discovery methods. With the rise of Python and its extensive libraries, virtual screening has become more accessible, efficient, and scalable. In this article, we will explore how Python transforms virtual screening in drug discovery, highlighting the core tools, processes, and benefits of using this versatile programming language.
What is Virtual Screening in Drug Discovery?
Virtual screening (VS) is a computational technique used to search large databases of small molecules to identify potential drug candidates that may bind to a target protein. It can be classified into two primary approaches:
- Structure-Based Virtual Screening (SBVS): This method relies on the 3D structure of the target protein, using techniques like molecular docking to predict how small molecules will bind to the protein’s active site.
- Ligand-Based Virtual Screening (LBVS): When the structure of the target protein is unknown, LBVS uses the known active ligands (drug-like molecules) to find compounds with similar structures or chemical properties.
Computer-aided drug discovery relies heavily on virtual screening to reduce the number of compounds that need to be tested in vitro, thus streamlining the drug development process.
The Role of Python in Virtual Screening in Drug Discovery
Python has revolutionized computer-aided drug discovery by offering powerful libraries and frameworks that enable researchers to automate and enhance virtual screening workflows. Its simplicity, flexibility, and extensive ecosystem of scientific libraries make it ideal for handling the complex tasks involved in screening drug candidates.
Several Python libraries and tools are essential for performing virtual screening in drug discovery, including:
- RDKit: For cheminformatics and molecular manipulation.
- PyMOL: For 3D visualization of molecular structures.
- AutoDockTools: For molecular docking.
- Open Babel: For converting and handling chemical file formats.
- scikit-learn and TensorFlow: For applying machine learning models to screening data.
By leveraging these libraries, Python makes virtual screening accessible not only to experienced computational chemists but also to researchers from various disciplines, speeding up the discovery of new therapeutic compounds.
Key Steps in Virtual Screening Using Python
1. Preparing the Compound Library
The first step in virtual screening is building and preparing a compound library. This library is essentially a collection of chemical structures that will be tested against the target protein. In ligand-based virtual screening, compounds with known biological activity are often included in the library.
Python’s RDKit library is widely used to manipulate, filter, and analyze chemical structures. RDKit can read a variety of file formats (e.g., SDF, SMILES) and calculate molecular properties such as molecular weight, solubility (LogP), and toxicity.
2. Molecular Docking
Molecular docking is a technique used in structure-based virtual screening to predict how a small molecule (drug candidate) will bind to a target protein. It involves placing the ligand in the active site of the protein and evaluating how well it fits, typically by calculating a binding affinity score.
Python’s AutoDockTools provides an interface for running molecular docking experiments using AutoDock, a widely used docking software. This allows users to automate the docking process, run multiple simulations, and analyze the results—all from within a Python script.
This process can be scaled to screen thousands of compounds against a protein target, making it a core component of structure-based virtual screening. Once docking is complete, researchers can rank the compounds based on their predicted binding affinity scores and select the top candidates for further study.
3. Molecular Fingerprinting and Similarity Searches
In ligand-based virtual screening, the goal is to identify compounds with similar chemical features to a known active ligand. Python makes this task easier with tools like RDKit, which can compute molecular fingerprints—binary representations of a molecule’s structure. These fingerprints are then used to calculate similarity scores between molecules.
By identifying compounds with high similarity scores, researchers can focus their efforts on molecules that are more likely to exhibit the desired biological activity.
4. Applying Machine Learning in Virtual Screening
Machine learning (ML) has become an essential part of computer-aided drug discovery, especially for analyzing and interpreting the large datasets generated during virtual screening. Python’s scikit-learn, TensorFlow, and PyTorchlibraries make it easy to build, train, and apply machine learning models to predict the biological activity of new compounds.
Machine learning models can quickly analyze large compound libraries and predict which molecules are most likely to succeed as drug candidates. This greatly accelerates the virtual screening in drug discovery process and improves the chances of identifying promising hits.
5. Analyzing and Visualizing Results
After completing a virtual screening campaign, analyzing the results is crucial for selecting the top drug candidates. Python offers numerous tools for visualizing molecular structures and results, such as PyMOL for 3D visualization and Matplotlib or Seaborn for plotting data.
With tools like PyMOL, researchers can visually inspect the binding interactions between a drug candidate and its target, providing deeper insights into the molecular basis of drug action.
Benefits of Python in Virtual Screening in Drug Discovery
The use of Python in virtual screening offers several key benefits:
- Automation: Python scripts can automate repetitive tasks, such as docking multiple compounds, processing large datasets, and filtering molecules based on desired properties.
- Reproducibility: Python code ensures that virtual screening workflows are reproducible and easily shared among researchers, fostering collaboration and accelerating drug discovery efforts.
- Scalability: Python’s versatility allows it to handle small-scale academic projects as well as large-scale industrial screening efforts.
- Integration: Python can integrate with various computational chemistry tools and databases, making it a comprehensive platform for computer-aided drug discovery.
Conclusion
Python has transformed virtual screening in drug discovery, offering powerful tools for automating, analyzing, and optimizing the drug discovery process. From molecular docking to machine learning, Python’s rich ecosystem of libraries and frameworks makes it easier for researchers to identify promising drug candidates quickly and efficiently.
If you want to explore more about applications of Introduction to Computer Aided Drug Discovery with Python you can join us in Bhopal for an exciting 2.5 Day Masterclass. More information is available HERE